1. 背景
转录组指的是生物细胞中所有转录物的总和,其可包括转录本的结构、功能和表达等方面。自人类基因组计划以来,解析基因组的功能,成为后基因组时代(功能基因组)必然的要求,尤其是在转录组分析方面,其能够在转录水平,通过衡量RNA分子的含量以评估基因的表达水平,以实现解析基因组或基因的功能。
现如今,转录组测序技术已经成为探究不同时空条件下的某种组织或细胞的基因在转录本层面的表达情况的一种重要手段。因而,在此简要概述转录组分析的基本流程。因作者水平有限,有纰漏之处,还请指教!
2. 转录组数据分析基本知识
2.1 差异倍数(fold change)
定义:fold change翻译过来就是倍数变化,假设某一基因在A样本中表达值为1,在B样本中表达值为3,那么B的表达就是A的3倍。一般我们都用count、TPM或FPKM来衡量基因表达水平,所以基因表达值肯定是非负数,那么fold change的取值就是(0, +∞).
为什么要用比值而不是差呢:假设A表达为1,B表达为8,C表达为64;直接用差B相对A就上调了7,C就相对B上调了56;用log2 fold change,B相对A就上调了3,C相对B也只上调了3. 通过测序观察我们发现,不同基因在细胞里的表达差异非常巨大,所以直接用差显然不合适,用log2 fold change更能表示相对的变化趋势。
缺陷:虽然大家都在用log2 fold change,但显然也是有缺点的:
(1)到底是5到10的变化大,还是100到120的变化大?
(2)5到10可能是由于技术误差导致的。所以当基因总的表达值很低时,log2 fold change的可信度就低了。

那么,对于其存在的缺陷,就可能需要统计学方法来评估其可信度了。尽管log2(FC)并不是后续假设检验中的统计量,但在一定程度上也能够反映出其可靠性。
2.2 相关的统计检验方法
我们在统计学中都知道,对于两个独立样本的均值比较问题,可以考虑利用t检验(若方差不等,则用welch t-test)进行假设检验。因此,我们是否可以直接利用t检验对差异表达基因进行分析呢,答案是不一定。在早期的针对基因表达进行高通量定量时,人们常常考虑利用相对低廉的基因芯片进行分析。基因芯片的荧光信号由于服从正态分布,因而可以直接利用t检验进行分析。
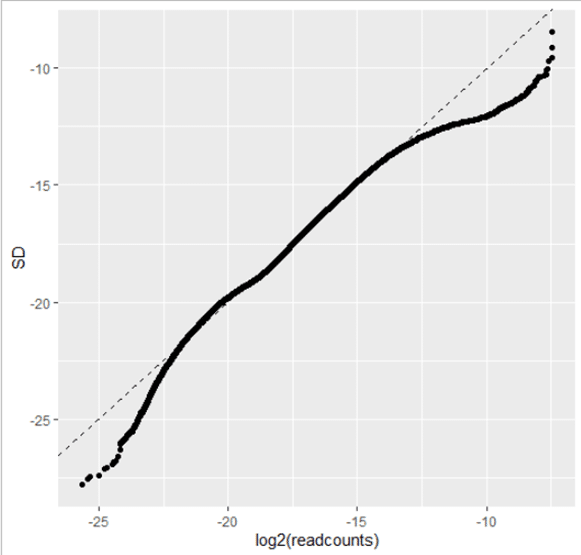

然而,在如今二代测序的成本已经下降到很低的程度,转录组测序早已成为一种常规的分析手段。然而,由于转录组数据是对读长(reads)进行定量,而reads数并不服从正态分布,而是泊松分布(后续确定为负二项分布)。由下图可以看出,其转录组测序的表达数据的方差并不是保持在一个恒定值,而是与均值呈现正相关关系。因此,对其进行差异表达的假设检验,并不能够简单地依赖于t检验。
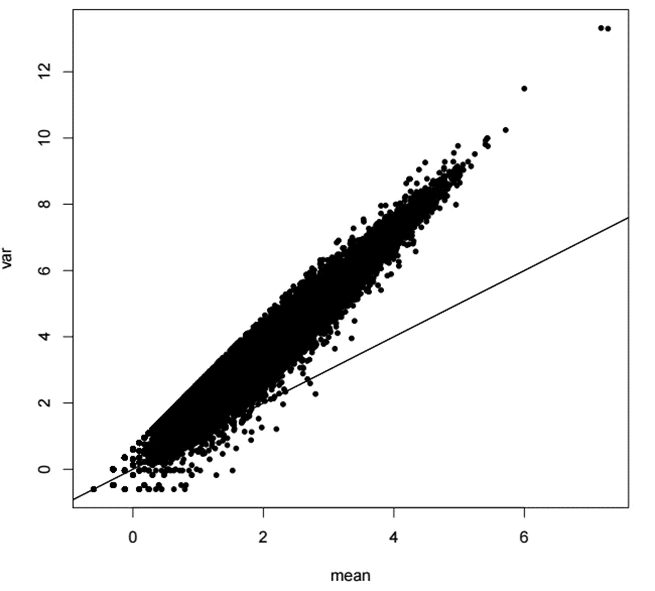
那么,该构造什么样的统计量进行,统计分析呢?这就涉及到了对表达数据进行标准化的方法了。
2.3 表达数据的标准化
最经典的基因标准化数据莫过于以下三种指标:RPKM、FPKM、TPM。RPKM,FPKM,TPM是为了消除基因长度和测序深度的影响。
在RNA-Seq的分析中,为了获得差异表达基因,只需要对不同基因的测序Read数进行比较即可。然而比对到不同基因上的Read数目并不能直接用于比较这两个基因的表达量差异,因为在RNA-seq中有一个很浅显的道理,基因越长,比对到此基因上的Read就会越多;测序深度越大,那么本次RNA-seq的所有Read数都会增加。也就是说Read数除了和基因表达量相关外,也和基因的长度、测序深度有关,因此为了比较多个RNA-seq重复(测序深度有一定差异)的不同基因(基因长度有一定差异)之间的表达量差异,那么就不能使用Read数直接进行比较,而是需要对Read数进行标准化。
RPKM: Reads Per Kilobase of exon model per Million mapped reads
- 计算总Read数:计算每一个RNA-seq样本的总Read数,然后将其换算为以百万位单位(M);
- 标准化总Read数:将所有基因的Read数除以总Read数;
- 标准化基因长度:再将所有基因的Read数除以基因长度(基因长度单位为kb);
$$RPKM=\frac{Reads/length(kb)}{Mapping reads/10^6}$$.
FPKM
- 其实FPKM同RPKM是一样的,只是RPKM用于单末端测序,而FPKM用于双末端测序。
- 二代测序时,会将所有的DNA打成片段(fragment),然后再去测序。单末端测序时,一个片段对应一个Read,但是双末端测序时,一个片段会从两端分别测定一次,因此这两个配对Read对应的是同一片段(偶尔也会有一个片段只对应一个Read的情况,另一个Read因为某些原因被剔除或丢失了)。
- 区别也就在这里,对于FPKM来说,配对到同一片段上的两个Read只会算作一个Read,也就是说FPKM是以Fragment为准,不以Read数为准,其他计算方式是完全一样的。
$$FPKM=\frac{Fragments/length(kb)}{Mapping fragments/10^6}$$.
TPM
TPM的计算方法其实同RPKM很类似,同样的对基因长度和测序深度进行标准化,只不过RPKM是先进行测序深度标准化,后进行基因长度标准化;而TPM是先进行基因长度标准化,后进行测序深度标准化。事实证明,TPM的标准化方法更有优势,为何会这样,见后述。这里先看看TPM的计算。
$$RPKM=\frac{Transcripts/length(kb)}{Mapping reads of transcripts/10^6}$$.
另外,还有更为可靠的标准化方法,包括:edgeR的TMM (trimmed mean of M-values),DESeq2的Median of Ratio。
2.4 常见差异表达分析R包
那么,有了标准化的方法,又该如何估计变化的离散程度(dispersion)呢?这就需要估计离散的数学模型,并且对基因范围的表达均值和离散程度进行拟合。常见的差异表达分析R包的标准化方法、拟合模型、离散估计以及假设检验的方法总结如下:
| DE analysis packages | Normalization | Distribution of readcounts matrix | Fitting | Dispersion assumption | Tests |
| Limma | NO (early version)/CPM (edgeR) | Normal/Negative binomial | GLM | eBayes/voom (mean-variance relationship) | Moderated t/F-test |
| edgeR | TMM (trimmed mean of M-values) (normalized by a gene set) | Negative binomial | GLM | eBayes/quantile-adjusted conditional maximum likelihood (single factor)/ Cox-Reid profile-adjusted likelihood (multiply factors) | Fisher’s exact test (single factor)/likelihood ratio tests & quasi-likelihood F-tests (multiply factors) |
| DESeq2 | Median of Ratio (normalized by one gene) | Negative binomial | GLM | maximum likelihood estimate/Cox-Reid adjusted profile likelihood | (nbinorm) Wald statistics/LRT test |
Limma
早期,Limma包主要针对基因芯片数据进行分析,直接利用基因表达的信号数据,因而没有标准化方法,而后续为了适用于转录组测序数据的分析,增添了CPM的标准化方法,并且利用了voom算法对均值和离散之间的关系进行拟合,估计其离散程度。
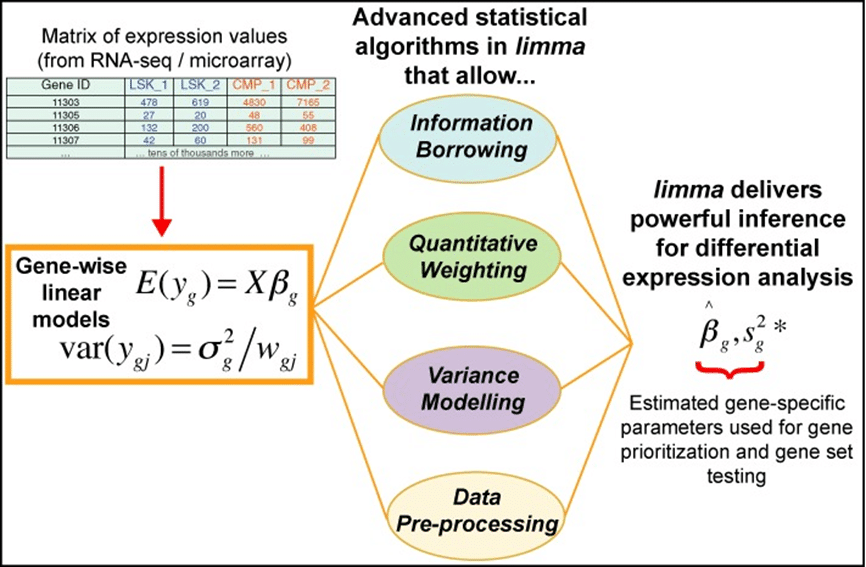
主要流程:
- lmFit():构建GLM (generalized linear model)拟合模型
- eBayes(): Dispersion (离散) 估计
- topTable():统计检验
edgeR
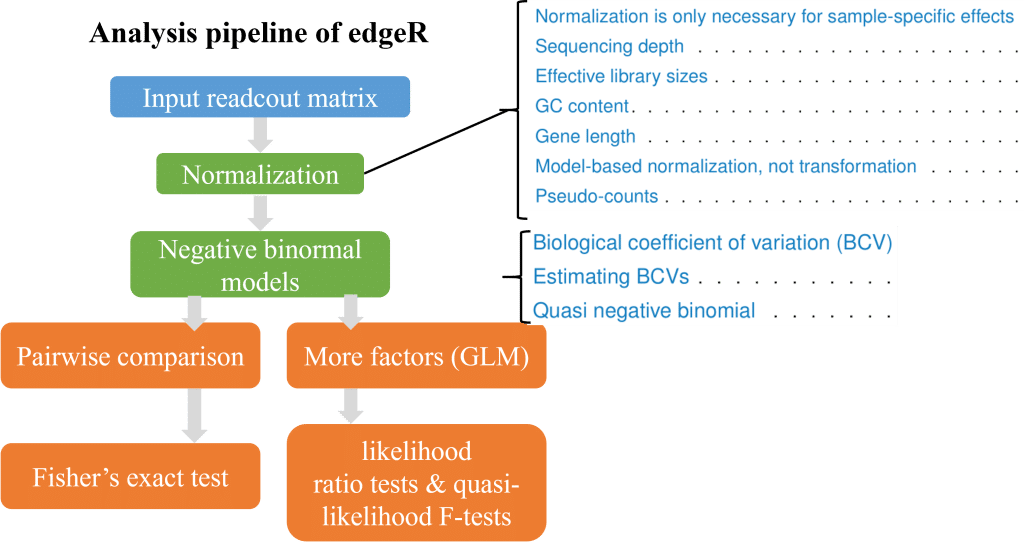
标准化:
- DGEList: Constructing expression list object
- calcNormFactors: Normalization
两组比较:
- estimateCommonDisp: calculating ordinary dispersion
- estimateTagwiseDisp: calculating gene-wide dispersion
- exactTest: Fisher exact test
多组比较:
- DGEList: Constructing expression list object
- estimateGLMCommonDisp: calculating GLM dispersion
- estimateGLMTrendedDisp: calculating gene-wide dispersion
- glmFit: Fitting
- glmLRT: Likelihood ratio test
DESeq2
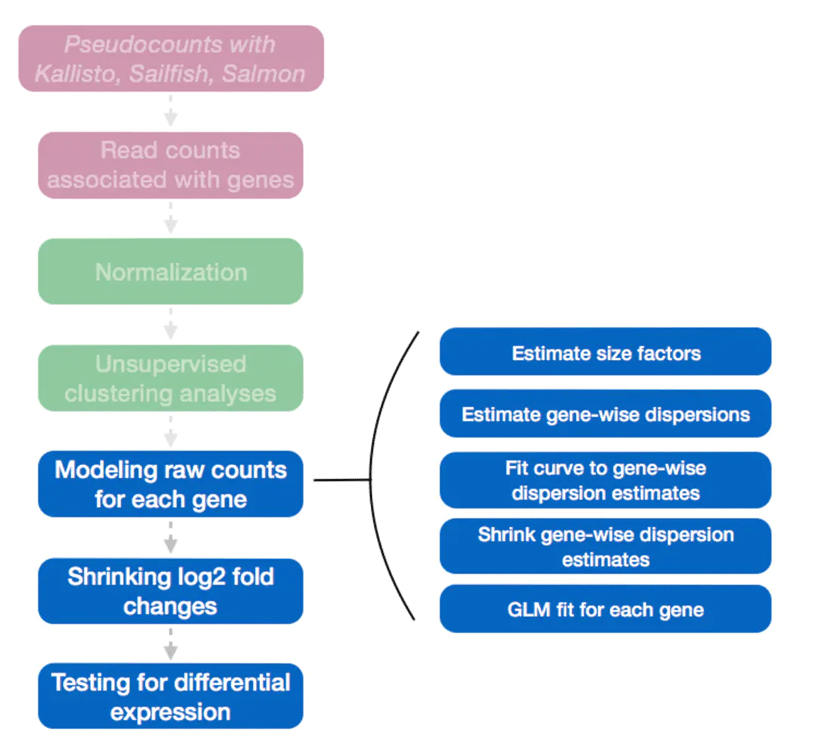
- DESeqDataSetFromMatrix: Constructing dds object
- DESeq:Normalization
- results:Differential analysis—wald test(one-factor)/LRT test (multi-factors)
下面,我们以DESeq2为例展示针对转录组数据的差异表达分析流程。
3. 以DESeq2为例的差异表达分析流程
3.1 准备基因表达矩阵数据(read counts)
DESeq2的输入表达数据可以有多个来源,如果是从以下几个基因/转录本定量工具得到的,则可以直接利用以下函数进行导入。
| function | package | framework | output | DESeq2 input function |
|---|---|---|---|---|
| tximport | tximport | R/Bioconductor | list of matrices | DESeqDataSetFromTximport |
| tximeta | tximeta | R/Bioconductor | SummarizedExperiment | DESeqDataSet |
| htseq-count | HTSeq | Python | files | DESeqDataSetFromHTSeq |
| featureCounts | Rsubread | R/Bioconductor | matrix | DESeqDataSetFromMatrix |
| summarizeOverlaps | GenomicAlignments | R/Bioconductor | SummarizedExperiment | DESeqDataSet |
首先,我们先导入相关的包,并导入上一步得到的表达矩阵:
## 加载所需的包
library(reshape2)
library(stringr)
library(dplyr)
library(ggplot2)
library(DESeq2)
# 运行前,先修改工作目录
setwd("/path/to/your/working/directory")
## Global configures
FILTER_GENE_NUMBER<-1
# FILTER_SAMPLE_NUMBER<-1
OUTPUT_DIR<-'./output/differential_expression_analysis'
FIGS_DIR<-'./figs'
# 创建输出目录(用于存储中间产生的必要结果数据和图)
if (!dir.exists('./output')){
dir.create('./output')
}
if (!dir.exists(FIGS_DIR)){
dir.create(FIGS_DIR)
}
if (!dir.exists(OUTPUT_DIR)){
dir.create(OUTPUT_DIR)
}
# 导入基因的表达数据
## Reading gene/transcript expression readcounts matrix
gene_expression_matrix<-read.csv('./Quantity/gene_count_matrix.csv',header = T)
head(gene_expression_matrix)
dim(gene_expression_matrix)
## Filtration by genes and samples
# discard genes with less than FILTER_GENE_NUMBER
col_number<-dim(gene_expression_matrix)[2]
gene_expression_matrix_filt<-gene_expression_matrix[
rowSums(gene_expression_matrix[,2:col_number]>0)>FILTER_GENE_NUMBER,]
# check how many genes were discarded
nrow(gene_expression_matrix)-nrow(gene_expression_matrix_filt)
# 2500 genes were discarded
可以看到,在过滤部分,我们过滤掉了2500个低表达的基因(表达的样本数)
3.2 基因表达过滤
在这里,我们也可以这对dds对象进行过滤和筛选,然而因为,我们在前面已经做了过滤,所以这部分就没有必要了。但是,记得要把基因的信息去掉,这里,我们把gene_id去掉,并将其作为行名(以保留该信息)。
之后,就可以构建DESeq对象。
# remove the first column gene_id
rownames(gene_expression_matrix_filt)<-gene_expression_matrix_filt$gene_id
gene_expression_matrix_filt<-gene_expression_matrix_filt[,-1]
# Main workflow of DESeq2
cts <- read.csv("./Quantity/gene_count_matrix.csv")
dds <- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design = ~ condition)
dds
## (Optional) Filtration of dds
# dds<-dds[rowSums(counts(dds)>1),]
# class: DESeqDataSet
# dim: 52298 80
# metadata(1): version
# assays(1): counts
# rownames(52298): MSTRG.15|CHRNA10 MSTRG.5 ... MSTRG.38152|LOC113840327
# MSTRG.38153
# rowData names(0):
# colnames(80): x x ... x
# colData names(4): x x x x3.3 基因表达过滤
之后,就可通过调用DESeq函数进行差异分析的工作流程:
- 估计标准化因子 estimation of size factors: estimateSizeFactors
- 估计离差 estimation of dispersion: estimateDispersions
- 拟合负二项分布GLM模型并做wald t检验 Negative Binomial GLM fitting and Wald statistics: nbinomWaldTest
# call DESeq function
dds <- DESeq(dds)
# get the analysis result
res <- results(dds)
# Process:
# estimating size factors
# estimating dispersions
# gene-wise dispersion estimates
# mean-dispersion relationship
# final dispersion estimates
# fitting model and testing
之后,我们就可以得到差异表达的结果了

